Integrating Microbiome and Clinical Data for Disease Risk Prediction in Cystic Fibrosis
This study developed a rule-based disease risk prediction model for cystic fibrosis patients that integrates microbiome composition, CFTR genetic variants, and clinical metadata using 16S rRNA sequencing data from public repositories. The model uses a baseline risk score of 50% with weighted adjustments for factors including CFTR mutations, clinical status, medications, age, and bacterial abundance to provide personalized risk assessments. Results demonstrated that therapeutic beta-lactam antibiotics significantly reduced microbiome diversity compared to subtherapeutic doses, correlating with increased exacerbation risk and highlighting the importance of preserving microbial diversity. This transparent, customizable framework advances precision medicine in cystic fibrosis management by providing clinicians with an evidence-based tool for individualized patient care.
STEM RESEARCHMEDICAL - HEALTHBIOMEDICAL INFORMATICSMICROBIOME RESEARCH
Jayesh Patil
7/3/202513 min read
Abstract
Cystic fibrosis (CF) exhibits significant inter-patient variability influenced by genetic factors, clinical history, and airway microbiome composition. This study developed a rule-based disease risk prediction model integrating microbiome data, CFTR genetic variants, and clinical metadata. Using 16S rRNA sequencing data from MicrobiomeDB.org, we analyzed bacterial taxa including Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cepacia alongside patient demographics and clinical measurements. The model uses a baseline risk score of 50% with weighted adjustments for CFTR mutations (homozygous +20%, heterozygous +10%), clinical status (untreated exacerbation +25%, recovery -10%), medications, age, and bacterial abundance. Results showed therapeutic beta-lactam antibiotics significantly reduced microbiome diversity compared to subtherapeutic doses, correlating with increased exacerbation risk. The model successfully identified high-risk CF patients by integrating microbiome and clinical parameters, demonstrating the importance of preserving microbial diversity. This transparent, customizable framework advances precision medicine in CF management, providing clinicians with a tool for individualized risk assessment based on genetics, microbiome composition, and clinical factors.
Keywords: cystic fibrosis, microbiome, CFTR mutations, disease risk prediction, personalized medicine, 16S rRNA sequencing, rule-based modeling
Introduction
The exploration of microbiome research has become an integral component of improving and expanding healthcare, specifically contributing to our understanding of how collective microbial communities interact with and affect human health. Specifically, in the context of cystic fibrosis (CF), this chronic condition will first be described and introduced as a genetic disease, which invites the question of its association with the resident microbial communities and how its interactions affect the risk of CF disease. The primary aim of this study is thus to determine how the microbiome composition and previous clinical history of a patient can be utilized in conjunction to predict the disease risk using a rule-based model. Patient microbiome and clinical metadata were obtained from MicrobiomeDB.org, a public repository that provides curated datasets from microbiome studies. Data used in this study included CFTR mutation type, clinical status, and microbial composition. To achieve this, the study employs 16S rRNA sequencing for precise bacterial taxon identification, including comprehensive clinical metadata for the patient samples. The convergence of these components may further enhance the ability to predict disease risk assessment and ultimately contribute toward more personalized treatments of cystic fibrosis.
Dataset Overview and Methodology
The implementation of 16S rRNA sequencing serves as a foundational tool for identifying bacterial taxa, allowing for the accurate determination of microbiome composition. This approach of sequencing the 16S ribosomal RNA gene is utilized to determine available unique sequences classified at different taxonomic levels, including phyla, classes, orders, families, genera, and even species (Huang et al., 2019, p. 142). In this study, the data derived from the “EcoCF_16S rRNA_(V4)_assay” are detailed and used as evidence to identify and characterize the respective bacterial taxa. Using an exhaustive method as such ensures that the analysis remains accurate, as these markers are specific to most bacteria and their variation is compared between species to ascertain relative assignments (Regueira-Iglesias et al., 2023). Upon sequencing and identifying the respective microbial communities through the 16S rRNA targeted sequencing, specific community characteristics can be associated with certain clinical outcomes of the patients highlighted in the study. This detail is invaluable as it allows these characteristics to be linked with the disease process specific to the organism in cystic fibrosis.
Additionally, participant metadata from the “EcoCF_Participant” sheet offers disease risk models an important background due to the array of demographic and genetic information that these diseases divulge. Notably, variants and mutations of the CFTR gene, which are associated with cystic fibrosis pathophysiology, are significant to personalized medicine approaches and reveal genetic characteristics associated with cystic fibrosis-specific ailments (Bradbury, 2021, p. 583). The crucial demographic information, such as participant sex and country of origin are critical variables that significantly affect the onset and progression of conditions associated with cystic fibrosis. When combined with available 16S rRNA sequencing data, participant metadata facilitates precise risk models through potential variations due to differences in genetic backgrounds and environmental exposure. Thus, the integration of these various data types supports a markedly detailed view of the disease, critical in formulating robust and targeted cystic fibrosis treatment plans for affected individuals.
Furthermore, the “EcoCF_participant_repeated_meas” sheet makes a significant contribution to the offer of longitudinal clinical metrics, such as the clinical visit number, age, and changes in medication over time. These parameters elaborately characterize clinical information for each particular patient and further emphasize the dynamic nature of cystic fibrosis treatment. The study analyzes the relationships of variations in clinical parameters and microbiome composition, which refines risk prediction assessments. Linking with participants supports the accuracy of bacterial taxa assignment and improves disease risk prediction performance and model interpretability (Zhao et al., 2021). The EcoCF_Sample sheet is a vital part of this study to connect the clinical and microbiome sample data with participants, as taxonomic labeling is employed for accurate statistical analyses. The presence and abundance of specific bacterial species in the cystic fibrosis airway microbiome, such as Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cepacia, are closely linked to disease severity and progression, making them key indicators for predicting individual disease risk.
Table 1
Common bacterial species isolated from the CF airway microbiome and their clinical significance in disease progression.
Microorganisms
Description and Clinical Significance in Cystic Fibrosis
Pseudomonas aeruginosa
P. aeruginosa is a dominant opportunistic pathogen in CF, known for its biofilm formation and antibiotic resistance, contributing to chronic pulmonary infection and progressive lung damage.
Staphylococcus aureus
S. aureus, including methicillin-resistant strains (MRSA), is frequently isolated in pediatric CF patients and is associated with increased airway inflammation and early structural lung changes.
Haemophilus influenzae
H. influenzae is commonly detected in younger individuals with CF and can contribute to acute exacerbations and persistent airway colonization.
Burkholderia cepacia
B. cepacia complex species exhibit high intrinsic antibiotic resistance and are associated with rapid pulmonary decline, increased hospitalization, and reduced transplant eligibility.
Microbiome Composition and Clinical Outcomes
Risk prediction in cystic fibrosis microbiome also relies on the impact of antimicrobial treatment, specifically beta-lactam antibiotics. This can be seen in the study, which revealed that beta-lactam antibiotics at therapeutic doses significantly impacted the microbial diversity within the microbiome than subtherapeutic doses (Hahn et al., 2019). As such, the level of microbial diversity significantly influences the clinical status of patients, for the loss of microbial diversity within the microbiome is associated with an increased disease state and pulmonary exacerbation risk. In particular, the overgrowth of Pseudomonas aeruginosa, an opportunistic pathogen that often becomes dominant when diversity declines, has been strongly linked to chronic infection and worsening lung function in cystic fibrosis patients. Therefore, the patterns of the microbiome composition impacted by antibiotic treatment in patients are essential in determining the precise risk prediction of treatment outcome in cystic fibrosis patients. The study also pointed out that the pattern observed in the microbial composition of the antibiotic therapy should be considered in the treatment prescription process, for the risk prediction involves optimizing drug treatment to account for loss of microbial diversity (Hahn et al., 2019).
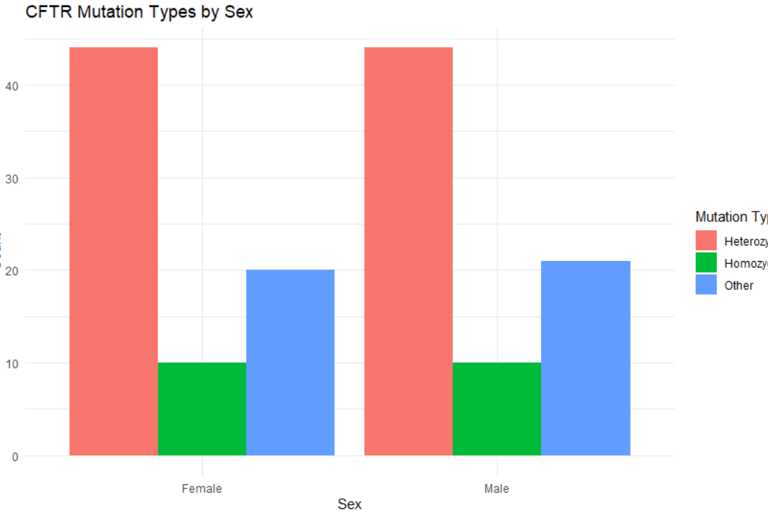
Figure 1
Distribution of CFTR mutation types by sex in individuals with Cystic Fibrosis. A higher prevalence of ΔF508 homozygous mutations was observed in males, while females showed a more diverse mutation profile.
Moreover, the study evaluating therapeutic and subtherapeutic beta-lactam antibiotic treatment in children with cystic fibrosis allows substantial insights into the microbiome's diversity transformations (Hahn et al., 2019). The findings demonstrated that treatment induced an exponential drop in the respiratory tract microbiome's diversity, which correlates with higher chances of pulmonary exacerbations. In contrast, the subtherapeutic treatment resulted in stable diversity, which relates to the lower influence on immediate clinical outcomes. These two scenarios illustrate the potential of antibiotic treatment strategies to prevent high-risk microbiomes' diversity shifts, and eventually prevent disease aggravation states and support a steady clinical course. Therefore, antibiotic treatment strategy in cystic fibrosis targeting diversity preservation requires thorough attention to achieve greater impact on clinical outcomes and refine disease risk dynamics prediction phenomena (Hahn et al., 2019).
Disease Risk Model: Design and Logic Explanation
The development of a manually weighted rule-based scoring system begins with a critical step in cystic fibrosis risk assessment, with targeted adjustments integrated to account for genetic factors and microbial profiles linked to disease progression. CFTR mutation types play a substantial role in the scoring model. Variations such as homozygous and F508del mutations can be meticulously included to increase or decrease the baseline risk score of 50%. CFTR mutation type influences disease expression, and in this manner, the approach must be unique, as it will be weighted following the influence the specific mutation type has over clinical progression and disease severity. Our approach is similar to that offered in the FARE model, where quantitative and qualitative information is crossbred into a logical rule-based system (the model) to produce the adverse event risk estimates (Cattelani et al., 2020). The accuracy of this method allows clinicians to make further risk-increasing or decreasing adjustments, all in the name of making a personalized risk estimate that is critically and specifically aligned with the individual genetic makeup variant. This represents a valuable step forward that only enhances the clinical application and impact of the model.
Code Block 1
Logic for adjusting disease risk score based on CFTR mutation type and clinical status. Risk values increase for homozygous mutations and untreated exacerbations, and decrease during recovery phases.
if (patient.getCftrMutationType().equalsIgnoreCase("Homozygous")) {
diseaseRisk += 20;
} else if (patient.getCftrMutationType().equalsIgnoreCase("Heterozygous")) {
diseaseRisk += 10;
}
if (patient.getClinicalStatus().equalsIgnoreCase("Exacerbation (no treatment)")) {
diseaseRisk += 25;
} else if (patient.getClinicalStatus().equalsIgnoreCase("Recovery (within 1 month)")) {
diseaseRisk -= 10;
}
In addition to the CFTR mutation type, the model integrates a series of clinically relevant variables that further refine the disease risk score. Beginning from a baseline risk of 50%, the score is adjusted using a series of weighted rules. If a patient presents with a homozygous CFTR mutation, 20% is added; if heterozygous, 10% is added. Specific mutation variants such as F508del and N1303K add 15% and 12% respectively. Medication history is factored in to reflect ongoing treatment effects: Azithromycin, Aztreonam, Dornase, and Tobramycin each reduce the score by 5%, while Colistin reduces it by 10%. Clinical status at the time of assessment contributes heavily: an untreated exacerbation results in a 25% increase, a treated exacerbation adds 15%, while being in recovery (within one month of completing antibiotics) reduces risk by 10%, and being on antibiotics without an exacerbation lowers it by 5%.
Code Block 2
This code adjusts the disease risk score based on clinical status and compares the patient’s median value to the average of upper and lower quartiles, increasing or decreasing risk accordingly.
if (patient.getClinicalStatus().equalsIgnoreCase("Exacerbation (no treatment)")) {
diseaseRisk += 25;
} else if (patient.getClinicalStatus().equalsIgnoreCase("Recovery (within 1 month)")) {
diseaseRisk -= 10;
}
//. . .
double avgQuartile = (patient.getUpperQuartile() + patient.getLowerQuartile()) / 2;
if (patient.getMedian() > avgQuartile) {
diseaseRisk += 5;
} else if (patient.getMedian() < avgQuartile) {
diseaseRisk -= 5;
}
Moreover, the scoring model uses the adjustments for medication, which is the key instrument to improve the risk evaluation efficacy in the cystic fibrosis population. Various medications, such as pancreatic enzyme replacements or CFTR modulators, are continuously integrated into the scoring model to assess the risk regarding their fundamental impact on the disease course and clinical outcomes in this population. Several medications can modify the clinical course, leading to alterations in the baseline risk score and requiring its recalibration based on the medication dosage and time exposure (Cattelani et al., 2020). Age is another variable, as patients under 12 have 5% subtracted from their score, while those over 40 have 5% added. Visit frequency is also considered: more than 10 visits add 5%, while three or fewer subtract 5%. The model also draws on microbiome-derived variables: a minimum bacterial abundance value below 50 leads to a 10% reduction, while a maximum above 500 leads to a 10% increase. Besides, the adjustments for clinical status and age represent key elements of the scoring model since they create additional background to the patient’s context prompt and disease stage. The above-mentioned criteria allow the scoring model to estimate the age-dependent risk differences and clinical findings, which improves its flexibility and coverage, making it more applicable in clinical practice.
Moreover, the logic of a rule-based scoring model is its provision of a flexible and transparent framework, constituting a coherent approach for combining various evidence sources. Like the healthcare domain FARE model (framework for adverse event risk) (Cattelani et al., 2020), this model allows for assigning a specific weight to each risk factor, which could be adjusted alongside the disease progression. In cystic fibrosis, pathologies involving a dynamic interplay of genetic, microbiome, and clinical factors can significantly benefit from the rule-based approach as the independent risk score weight can be tailored according to the indices sourced from qualitative and quantitative evidence, thus enabling more accurate prediction. Additionally, the impressive flexibility of the rule-based model promotes the provision of frequent updates, as findings regarding the clinical or microbial aspects become known, ensuring the continuous adjustment of the disease risk prediction algorithm. Therefore, taking into account provided attributes and characteristics, the scoring model is flexible enough to adapt under the specific disease and individual patient scenario, while also providing solid application where disease risk is concerned, thus positively impacting the cystic fibrosis course prediction and individualized treatment strategies (Zhao et al., 2021). This approach is further exemplified in the specific application of bacterial abundance adjustments within the model. If the sample's median bacterial abundance exceeds the average of its upper and lower quartiles, 5% is added; if it's lower, 5% is subtracted. Once all of these adjustments are applied, the final risk score is bounded between 0% and 100%, ensuring it remains interpretable and clinically meaningful. Through this multi-layered, rule-based architecture, the model delivers individualized disease risk estimates that are directly shaped by the patient’s genetic profile, clinical context, and airway microbiome composition.
Data Interpretation and Implications
The application of the model results indicates that it is possible to recognize the high-risk patients suffering from cystic fibrosis with the help of precise mapping of the integration of data on the microbiome and clinical metadata. This finding highlights the ability of modeling to predict with an impressive result when there is convergence of information on nonpathogenic taxa and pathogen data available in the model, and it improves the predictive performance of the models for lung function (Zhao et al., 2021). The models help to evaluate the treatment plan tailored according to an individual risk profile. The rule-based model is designed transparently, and it can be customized. Clinicians have an opportunity to customize the model and adjust the modeled risk profile of each patient as the data set regenerates to ensure treatment attention is personalized concerning the changes in the composition of the microbiome profile and status of the disease. Customization enhances the performance of the patient risk-adjusted treatment plan; furthermore, it allows for establishing a mechanism for improving the model and its performance concerning the management of the disease in future scenarios (Hahn et al., 2019).
Regardless, there are certain limitations in the predictive model that should be considered to improve its predictive value despite having a well-structured framework. One major limitation is the microbiome composition variation due to various factors such as diet and lifestyle, environmental exposure, and other comorbidities. These factors can lead to discrepancies in data (Chrisman et al., 2021). Furthermore, although the present model utilizes 16S rRNA sequencing and clinical metadata, the interplay between the host genetics and environment may not be completely covered. This indicates the need for further studies to obtain more integrative biomarkers. Also, in a rule-based scoring model such as this, there might be different microorganisms that are emerging and may play a vital role in undocumented scenarios; therefore, continuous data updating of the model is necessary (Chrisman et al., 2021). Future studies may explore establishing more prediction parameters for microbial-specific clinical cases by utilizing advanced data integration methods such as sequence-based biomarkers.
Conclusion and Future Directions
The combination of predictive genetic, clinical, and microbiome data represents an innovative approach to personalized medicine, delivering an accurate platform for predicting associated disease risk in cystic fibrosis. With the establishment of a rule-based model structured to align with each of these heterogeneous datasets, practitioners could enhance risk prediction accuracy, thereby optimizing the care process. Due to the model's unique versatility, it could be continually improved by adapting new microbiome data and more precise biomarkers, achieving remarkable risk prediction success globally. Furthermore, the evolving nature of the model embraces its application in diagnostic models, paving the way to promising therapeutic strategies for more timely interventions in chronic diseases beyond cystic fibrosis. Therefore, the continuous evolution of the model ensures that future discoveries broaden its functionality and significantly impact the field of personalized healthcare and precision medicine.
To conclude, future studies involve integrating the use of more data types and innovative techniques to improve prediction models in cystic fibrosis treatment. It also calls for the use of analytical strategies that center on the combined sequencing of the genome and the host-microbe study. Such analyses have the potential to enhance the understanding of host-microbe communications and present new biomarkers that could help identify the patient risk of the disease (Regueira-Iglesias et al., 2023). The analytical strategy could also use the multi-omic-wide approach and include the data from metabolomic and transcriptomic analyses. These methods would further improve the understanding of microbiome function with disease outcomes. The study may also adopt other machine learning methods to integrate data from multiple sources and advance the predictive value of models that exist today. All these innovative approaches are needed to address the current gaps and push precision medicine so that they may translate into better therapeutic modalities and patient results in managing cystic fibrosis.
After filtering data based on the CFTR mutations, adjustments in the model should be made considering patient heterogeneity, predicted risk levels should remain accurate and relevant when a patient belonging to another group of the affected CFTR gene is identified (Bradbury, 2021, p. 592). Due to the heterogeneity of the clinical picture of each patient, a personalized approach is essential to secure the best fit of the model in the target population, and it constitutes another argument for the model's flexibility. Newly identified clinical and microbiome data should be integrated into the model to fit the best risk prediction to every specific subject or subgroup of patients.
References
Bradbury, N. A. (2021). CFTR and cystic fibrosis: A need for personalized medicine. In Studies of epithelial transporters and ion channels (pp. 547–604). Springer. https://doi.org/10.1007/978-3-030-55454-5_15
Cattelani, L., Chesani, F., Palmerini, L., Palumbo, P., Chiari, L., & Bandinelli, S. (2020). A rule-based framework for risk assessment in the health domain. International Journal of Approximate Reasoning, 119, 242–259. https://doi.org/10.1016/j.ijar.2019.12.018
Chrisman, B. S., Paskov, K. M., Stockham, N., Jung, J.-Y., Varma, M., Washington, P. Y., Tataru, C., Iwai, S., DeSantis, T. Z., David, M., & Wall, D. P. (2021). Improved detection of disease-associated gut microbes using 16S sequence-based biomarkers. BMC Bioinformatics, 22, 509. https://doi.org/10.1186/s12859-021-04427-7
Hahn, A., Fanous, H., Jensen, C., Chaney, H., Sami, I., Perez, G. F., Koumbourlis, A. C., Louie, S., Bost, J. E., van den Anker, J. N., Freishtat, R. J., Zemanick, E. T., & Crandall, K. A. (2019). Changes in microbiome diversity following beta-lactam antibiotic treatment are associated with therapeutic versus subtherapeutic antibiotic exposure in cystic fibrosis. Scientific Reports, 9, 2534. https://doi.org/10.1038/s41598-019-38984-y
Huang, Y.-A., Huang, Z.-A., You, Z.-H., Hu, P., Li, L.-P., Li, Z.-W., & Wang, L. (2019). Precise prediction of pathogenic microorganisms using 16S rRNA gene sequences. In Intelligent Computing Theories and Applications (Vol. 11644, pp. 138–150). Springer. https://doi.org/10.1007/978-3-030-26969-2_13
Regueira-Iglesias, A., Balsa-Castro, C., Blanco-Pintos, T., & Tomás, I. (2023). Critical review of 16S rRNA gene sequencing workflow in microbiome studies: From primer selection to advanced data analysis. Molecular Oral Microbiology, 38(5), 347–399. https://doi.org/10.1111/omi.12422
Zhao, C. Y., Hao, Y., Wang, Y., Varga, J. J., Stecenko, A. A., Goldberg, J. B., & Brown, S. P. (2021). Microbiome data enhances predictive models of lung function in people with cystic fibrosis. The Journal of Infectious Diseases, 223(Supplement_3), S246–S256. https://doi.org/10.1093/infdis/jiaa655
Appendices
Appendix A
public class DiseaseRiskModel {
public static void main(String[] args) {
PatientData patient = new PatientData();
double diseaseRisk = calculateDiseaseRisk(patient);
System.out.println("Patient's Disease Risk: " + diseaseRisk + "%");
}
private static double calculateDiseaseRisk(PatientData patient) {
double diseaseRisk = 50;
if (patient.getCftrMutationType().equalsIgnoreCase("Homozygous")) {
diseaseRisk += 20;
} else if (patient.getCftrMutationType().equalsIgnoreCase("Heterozygous")) {
diseaseRisk += 10;
}
if (patient.getCftrMutation1().equals("F508del") || patient.getCftrMutation2().equals("F508del")) {
diseaseRisk += 15;
}
if (patient.getCftrMutation1().equals("N1303K") || patient.getCftrMutation2().equals("N1303K")) {
diseaseRisk += 12;
}
if (patient.isAzithromycin()) {
diseaseRisk -= 5;
}
if (patient.isAztreonam()) {
diseaseRisk -= 5;
}
if (patient.isColistin()) {
diseaseRisk -= 10;
}
if (patient.isDornase()) {
diseaseRisk -= 5;
}
if (patient.isTobramycin()) {
diseaseRisk -= 5;
}
if (patient.getClinicalStatus().equalsIgnoreCase("Exacerbation (no treatment)")) {
diseaseRisk += 25;
} else if (patient.getClinicalStatus().equalsIgnoreCase("Exacerbation (treatment with antibiotics)")) {
diseaseRisk += 15;
} else if (patient.getClinicalStatus().equalsIgnoreCase("Recovery (within 1 month after stopping antibiotics for exacerbation)")) {
diseaseRisk -= 10;
} else if (patient.getClinicalStatus().equalsIgnoreCase("On Antibiotics No Exacerbation")) {
diseaseRisk -= 5;
}
if (patient.getAge() < 12) {
diseaseRisk -= 5;
} else if (patient.getAge() > 40) {
diseaseRisk += 5;
}
if (patient.getVisitNumber() > 10) {
diseaseRisk += 5;
} else if (patient.getVisitNumber() <= 3) {
diseaseRisk -= 5;
}
if (patient.getMin() < 50) {
diseaseRisk -= 10;
} else if (patient.getMax() > 500) {
diseaseRisk += 10;
}
double avgQuartile = (patient.getUpperQuartile() + patient.getLowerQuartile()) / 2;
if (patient.getMedian() > avgQuartile) {
diseaseRisk += 5;
} else if (patient.getMedian() < avgQuartile) {
diseaseRisk -= 5;
}
return Math.min(Math.max(diseaseRisk, 0), 100);
}
}